论文笔记:几何EGNN抗体CDR设计
Conditional Antibody Design as 3D Equivariant Graph Translation
文章: MEAN
代码: MEAN
会议:ICLR 2023 (Outstanding Paper Honorable Mention)

背景 Abstract & Introduction
CDR(Complementarity-Determining Regions)为抗原与抗体接触的地方,故又称抗原结合位。CDR为全身变异性最大的地方,以捕捉不同抗原,所以也常被称为高变区(hypervariable regions)。
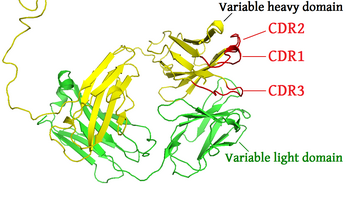
抗体设计对于生物学研究和临床治疗非常重要。现有的深度学习方法都面临以下几个问题:
- 生成CDR区域所用到的context(结构/序列)语境不充分
- 无法捕获输入结构的全部3D几何信息
- 使用自回归方法预测CDR序列计算复杂度太高
本文提出了一个多通道的等变注意力网络(Multi-channel Equivariant Attention Network, MEAN),可以同时设计抗体CDR的1D序列和3D结构。具体来说,MEAN将抗体设计视作一个条件图转换问题(conditional graph translation problem),通过引入目标抗原和抗体的轻链(light chain)。随后,MEAN使用E(3)等变的MPNN,结合注意力机制,可以同时捕获不同生物分子之间的几何关联。最终,MEAN通过多次迭代一次性输出1D的序列和3D的结构,相比于自回归方法,更加有效而且准确。MEAN方法显著优于当前1)序列和结构建模、2)抗原结合CDR设计、3)结合亲和力优化的SOTA方法,准确来说,相比于baseline,MEAN再抗体结合CDR设计上提高了23%,在抗体亲和力优化上提升了34%。
抗体我们免疫系统用于捕获特定病原的是Y形状蛋白。由于其特异性强,在临床治疗和生物学研究中凸显出重要的作用(每种抗体都可以特异结合到对应的抗原antigen上面)。而抗原-抗体结合区域则主要位于抗体的CDR区域,因此,抗体设计中一个重要问题即为找到可以结合到特定抗原的CDR序列,并且该序列拥有特定的性质,如较高的亲和力和胶质稳定性。许多工作利用深度生成模型来设计抗体,传统的方法只关注于建模1D的CDR序列,而最近的一个工作使用GNN来同时设计CDR的1D序列和3D结构。
尽管当前研究如火如荼,现有的方法对于建模抗体-抗原之间的空间关系仍尚有不足。首先一点,对于生物学语境的利用就是不充分的。先前的工作只关注于捕捉CDRs和同一个链上的蛋白质骨架结构信息,而忽略了目标抗原和抗体上其他链的结构信息,这一不足很可能会模型由于缺少完整的生物学信息而不能在抗体设计中表现得更好,如亲和力上面。此外,目前的工作不能够利用输入结构的全部3D几何信息。结构生物学的一个最重要的属性就是,每个结构(分子、蛋白质,等)都应该不受观察视角的影响(独立于观察视角),即E(3)等变性。为了解决这些限制,Jin等的方法预先将3D坐标处理为不变的特征后再传入模型,然而这样的预处理会导致结构信息如方向在特征和隐空间中丢失,从而使模型不能有效地学到抗原-抗体不同残基之间的空间相似性。除此之外,目前的生成模型都是一个一个预测氨基酸,这样自回归的范式也导致了模型推理的低效和推理误差的积累。
为了解决上述问题,本文将抗体设计表示为E(3)等变图转换(E(3)-quivariant graph translation),我们的贡献如下:
新的任务。利用条件生成,输入不仅包含CDRs重链的结构信息,也包括了抗原和轻链的信息。
新的模型架构。我们提出了端对端的多通道等变注意力网络(MEAN),可以同时输出CDRs的1D序列和3D结构。MEAN的算子直接在3D坐标空间中进行运算,并具有E(3)-等变性,不同于先前的E(3)-不变性,因此可以保持残基的全部几何结构。通过在内部语境编码器和外部注意力编码器中切换,MEAN可以利用3D信息传递(聚合+池化)与等变注意力机制来捕获抗原抗体复合体不同组件中的长距离、空间复杂的互作。
高效的预测。MEAN提出了通过几次迭代逐步生成CDRs的范式,而每次迭代都在序列和结构上进行完全的生成(full-shot)。这种逐步式(progressive)的完全的解码策略不会受限于像自回归那样的累计误差,并且在推理中更为高效。我们使用3个任务来证明我们方法的有效性:
- 序列和结构建模;
- 抗原结合表位CDR设计;
- 结合亲和力优化;
与先前的方法相比,我们这种不忽略轻链和抗原结构信息的方法,在以上3个任务中都达到了SOTA。
研究综述 Related Work
抗体设计Antibody design
以前的方法利用手工的能量函数来优化抗体,依赖于费时费力的分子模拟,而且由于天然结构中链链互作是极为复杂的生物学网络,所以利用力场和统计函数不能完全的建模这个生物学过程。因此,随着算力和深度学习的发展,大部分研究开始使用深度生成模型来预测更优的序列组合;最近,为了将3D结构整合进来,一些工作开始在序列和结构上同时建模,其利用距离矩阵将抗体表示为E(3)不变的特征,并自回归式地生成序列;之后的工作进一步整合了抗原表位(抗原上结合抗体的部分),但只考虑了CDR-H3,而忽略了抗体中的其它组件。不同于上述的工作,我们的方法考虑了更全面的信息,囊括1D和抗原抗体的3D信息,更重要的是,我们开发的E(3)等变模型非常适合于表示并建模3D结构之间的几何关系,而且不同于普遍的蛋白质设计,利用结构预测序列,我们的模型同时输出序列和结构。
等变图神经网络 Equivariant Graph Neural Network
自AlphaFold以来,在不同生物学领域的结构数据越来越多,从而很多几何等变的图神经网络应运而生来make sense这些结构数据。本文中,我们利用了标量化的E(n)等变GNN作为MEAN模型中的主要元件。具体来说,我们采用了多通道版本的等变图网络(改进自Huang et al.Equivariant graph mechanics networks with constraints),同时,我们开发了一种新型的等变注意力机制,可以更好的捕获抗体-抗原的关系。
方法 Method
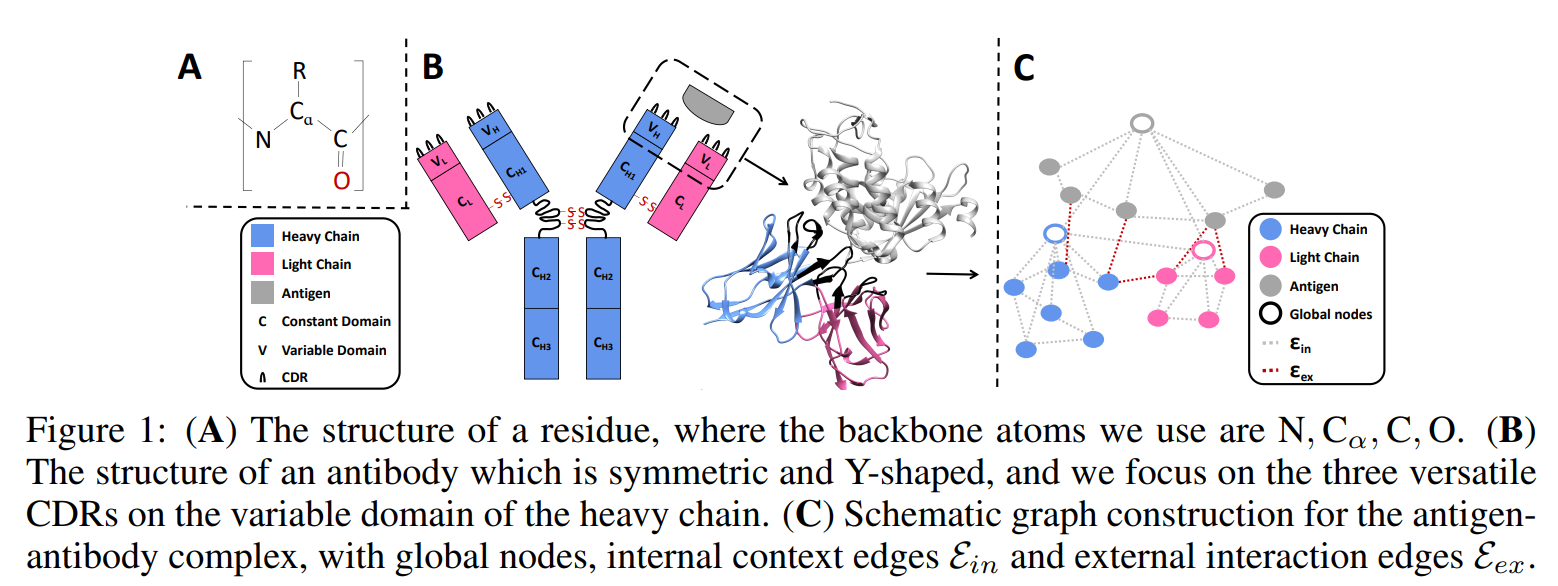
预备知识,符号, 建模
抗体是包含两个对称结构的Y形状的蛋白,每个结构由一个重链和一个轻链构成(图1)。在每个链中,包括许多保守的结构域,以及包括CDRs的可变结构域(
我们将每个抗体-抗原复合体的3个空间组分(抗原、轻链和重链)表示为图,表示为:
边构建 Edge construction
下面是构建边的细节。对于内部边来说,我们将
全局节点 Global node
CDR环的形状与抗体框架区的构想关系密切。因此,为了使生成的CDRs可以利用到其整个链的结构信息,我们进一步在每个组分中添加了一个全局节点,这个节点可以与这个组分中所有节点相连。此外,不同组分之间的全局节点都互相连接,而且与全局节点有关的所有边都位于
任务描述 Task formulation
已知3D抗体-抗原复合体图
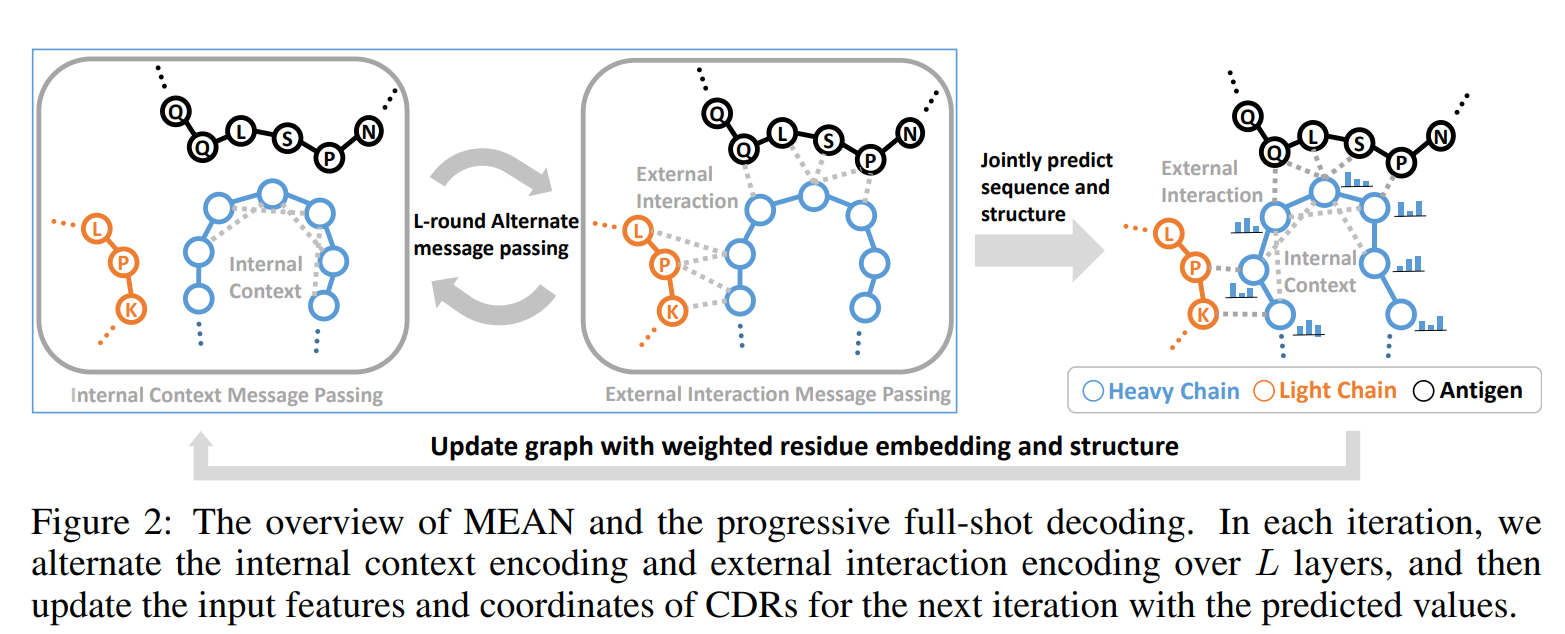
多通道等变注意力网络 MEAN
为了得到一个有效的转换器, 很重要的一点就是捕获不同链上残基的几何关系. 在E(3)等变GNN中的信息传奇机制可以很好得满足这一点. 特别地, 我们开发了MEAN来刻化输入复合体的几何和拓扑结构. MEAN的每一层都在不同的两个模块中转换(链内和链间), 即内部语境编码器和外部语境编码器, 这一想法是来自于一个生物学见解: 内部和外部残基之间的互作是各不相同的, 内部残基主要由于原子之间的相互作用力, 而外部主要来自于分子之间的作用力. 在几层信息传递后, 节点表示和坐标被转移到一个预测模块. 注意所有的模块都是E(3)等变的.
内部语境编码器 Internal context encoder
与GMN类似, 我们进一步将EGNN改进为多通道输入, 每一个残基都由多个骨架原子组成. 假设在
其中,
外部注意力编码器
这个模块利用图注意力机制来描述不同元件里残基之间的联系,但是不同于Velickovic et al,我们基于上述内部语境编码器中的多通道标量化设计了E(3)等变的图注意力机制。于是,我们有:
这里,
这些算子都是E(3)不变的,而且函数
输出模块 Output module
在
Theorem 1
将MEAN的转换过程表示为
于是
群算子
并且
逐步完全解码 Progressive Full-Shot Decoding
传统方法如RefineGNN使用自回归的方式来设计CDR序列,即以此只生成一个氨基酸。尽管这样的策略可以减少生成的复杂度,但是它难免会有一些计算上的额外开销,因此在长CDR序列上会由于梯度消失等问题难以训练;此外,自回归的方法随着推理次数的增多也会导致误差的积累。因此,多亏了MEAN强大的表达能力,我们可以通过
具体来说, 给定第
对于序列预测,每个节点都是监督学习,其交叉熵损失为:
对于结构预测,我们只在最后一个迭代执行监督。因为坐标数据中经常会有噪声,所以我们使用了Huber损失而不是平常的MSE损失,以此来避免数值不稳定性造成的差异(附录F中有详细解释):
最终,我们将上述两个损失平衡到一起
实验
我们在三个挑战任务上评估了我们的模型:1. 结构抗体数据库的生成任务(4.1);2. 从一个整理好的benchmark中收集的60个抗体-抗原复合体的抗体结合区域CDR-H3的设计(4.2);3. 突变蛋白互作(Mutant Protein Interactions)中结构动力学和能量数据库(Structural Kinetic and Energetic database)的抗原抗体结合亲和力的优化(4.3)。我们也提供了一个新的应用场景,即结合位置位置的情况(4.4)。
下面是我们选择的进行比较的baselines。第一,基于LSTM的模型来编码重链CDR结构语境,并输出CDRs序列。我们在编码器和解码器之间实现了cross attention机制,但是只使用了序列信息。此外,我们还进一步测试了C-LSTM来考虑抗原抗体复合体的全局信息,每个组分都被一个特殊的token分开。RefineGNN与我们的方法相近,它也在抗体生成过程中考虑了3D几何信息,但与我们不同的是它只是E(3)不变并且是自回归的方法。因为其原始版本只考虑了重链信息,我们进一步实现了全局信息版本的C-RefineGNN,具体来说,每个组分在序列中都被一个特殊token分开,在结构中加入了一个虚拟的节点。我们的模型命名为MEAN。每个模型训练了20个epochs,并且选择了loss最低的checkpoint在测试集上面测试,使用Adam优化器(学习率为1e-3)。对于MEAN来说,我们跑了3个迭代来解码。更多信息请看附录 D,LSTM和RefineGNN都按照其源码中的默认设置。
序列和结构建模
为了评估,我们使用氨基酸恢复率(Amino Acid Recovery, AAR),定义为预测序列和真实序列之间重叠的比率,以及两个结构之间的RMSD作为评价指标。多亏了模型的等变性,其能够直接计算坐标间的RMSD,不像其它的方法必须要使用Kabsch技巧来比对预测结构和真实结构后才能进行计算RMSD。我们的模型需要的输入必须完整,即复合体要包括重链、轻链和抗原三个元件。因此,我们从SAbDab数据库(Structural Antibody Database)中选择了3 127个复合体,并去除了缺少这些元件的结构。所有选择的复合体都按照IMGT重新进行了编号。我们根据CDRs的聚类来划分训练集、验证集和测试集。具体地,对每一种CDRs,我们首先用MMseqs2,其将抗体CDR区域相似度大于40%的聚为一类,这里采用BLOSUM62替换矩阵来计算序列相似性。对于CDR-H1,CDR-H2,CDR-H3的聚类数量分别为765,1093和1659。然后,我们按照8:1:1的比率将所有类都划分为训练、验证和测试集。我们使用了10倍交叉验证来获得最终结果,细节再附录A中。
结果
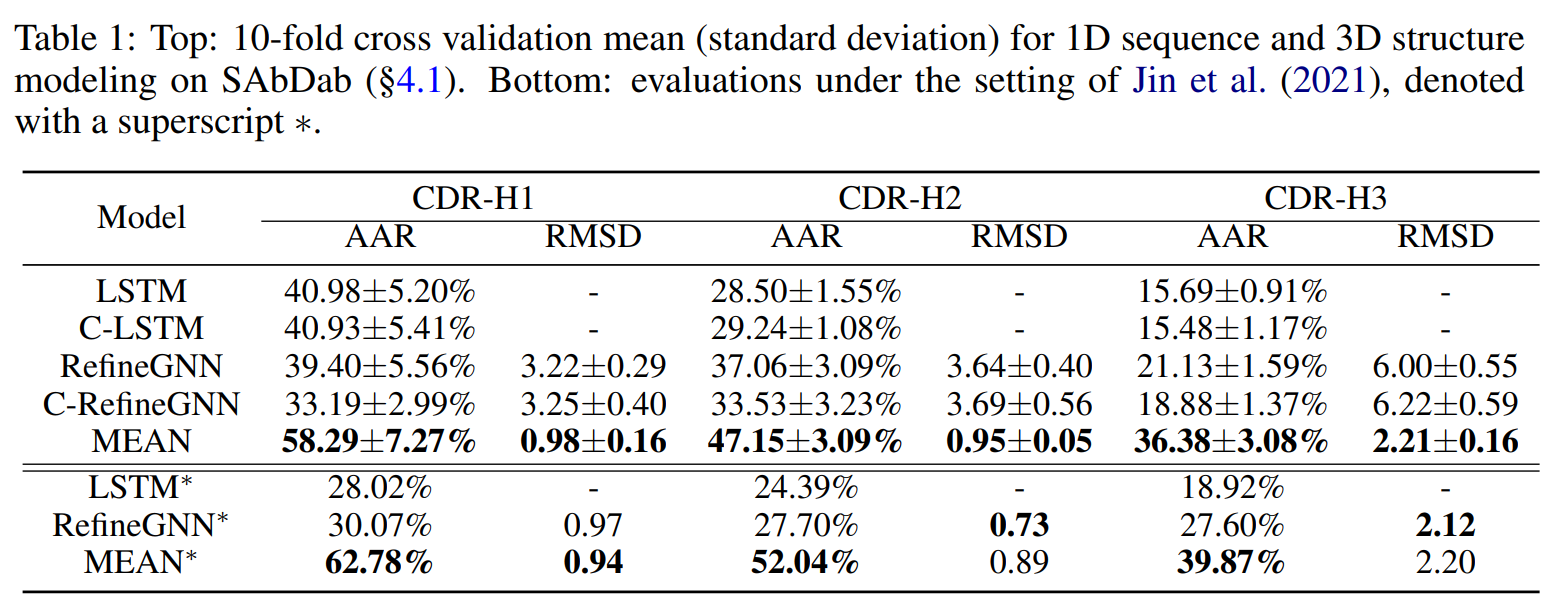
表1证明了MEAN方法在1D序列和3D结构建模中显著优于其它方法,这表明了MEAN在建模复合体潜在分布的有效性。当将考虑部分信息的LSTM/RefineGNN与考虑全局信息的C-LSTM/C-RefineGNN进行比较时,表明额外使用轻链和抗原信息导致性能下。这一现象说明如果CDRs和额外输入组件之间的相互依赖关系没有正确提供的话,模型的性能不会改进甚至降低。反之,MEAN在更多信息情况下性能更好,在第5节有消融实验证明了这一点。我们也在同样的数据集划分情况下比较了MEAN、LSTM和RefineGNN,表示为带有*的模型:MEAN*、LSTM*和RefineGNN*。具体地,LSTM*和RefineGNN*在全部数据集上训练,而MEAN只在完整的结构数据集上训练(大概占所有复合体结构的52%)。所有3个模型都在同样测试集上测试,即便如此,就AAR和RMSD两个指标而言,MEAN仍比其它两个方法性能更好,表明其强大的泛化能力。
抗原结合的CDR-H3设计
对于结合抗原的CDR-H3的设计,我们进行了深入的验证。除了AAR和RMSD,我们还加入了TM-score指标,其旨在计算两个结构全局相似度(为0到1之间),以此来评估设计的CDRs和框架结构是否相适合。我们使用官方的实验来计算TM-score:实现TM-score。对于自回归的基线,根据Jin et al. 的研究,为了减少误差积累,我们生成了10000个CDR-H3s,并选择了PPL最低的100个作为评估。值得注意的是,利用逐步完全解码策略,MEAN抗原大大减少误差的积累,因此可以直接生成目标CDR-H3。我们也将RosettaAD加入了基线中进行比较,这是一个基于传统方法的广泛使用基线。我们比较了所有方法在60个复合体上的性能,训练仍是使用SAbDab数据集,但我们移除了所有与benchmark数据集中60个复合体中CDR-H3聚在同一类的训练数据,以此避免任何可能的数据泄露。我们将剩余的结构分为9:1的训练集和验证集,两个数据集上的聚类/抗体数量分别为1443/2638和160/339。
结果
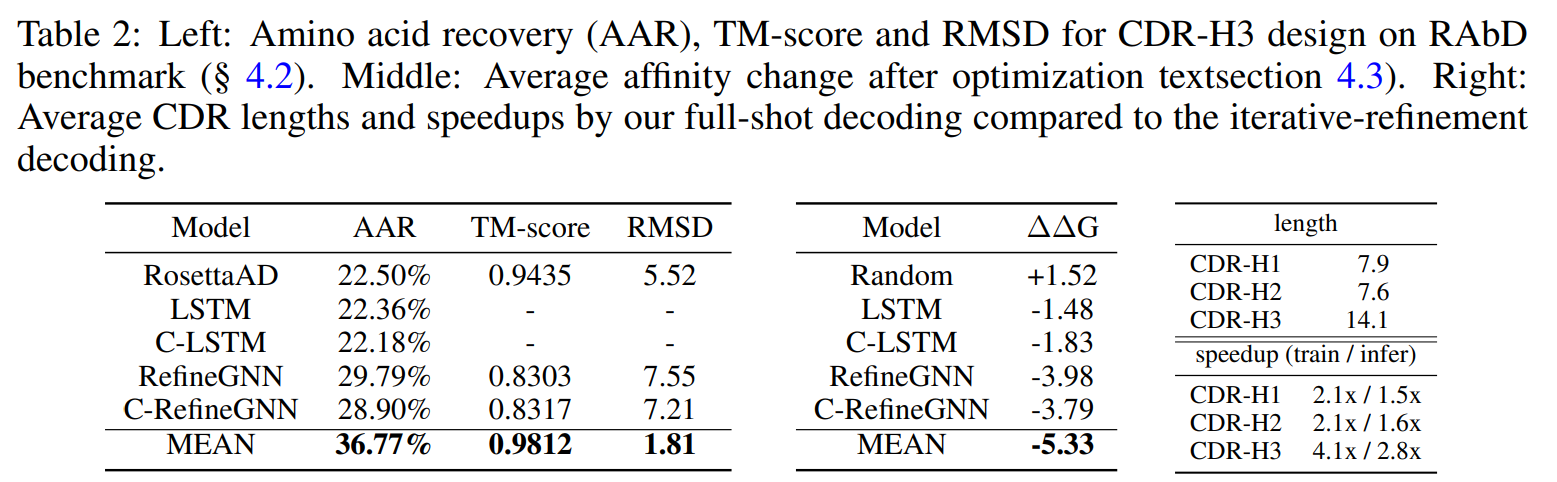
表2表明MEAN比其它方法在AAR和TM-score指标上性能更好。尤其是TM-score,MEAN可以达到0.99,表明设计的结构和原先的基本上一样。为了更好的展示这个效果,我们在图3中可视化了这个比较,MEAN生成的结构几何与原始结构重合,而RefineGNN存在一些偏差。
亲和力优化
对于疾病治疗来说,优化抗体的属性如亲和力是一个极为重要的任务。这可以表示为一个搜索问题,即在生成模型的结果中进行择优。在我们的例子中,我们同时优化了CDR-H3的序列和结构,以此来改进任何给定抗原抗体复合体的亲和力。为了评估,我们使用Shan et al. 中的几何网络来预测优化后结构的结合自由能 ΔΔG,具体地,我们利用SKEMPI V2.0的官方实现。ΔΔG的单位是kcal/mol,ΔΔG值越低说明亲和力越好。因为所有方法都只建模了骨架结构,所以在亲和力预测前我们使用Rosetta来对侧链进行选择。为了保证预估的泛化能力,我们从训练集(SKEMPI V2.0)中选择了53个抗体。此外,我们将SAbDab按照9:1的比例分为训练集:验证集来预训练模型。
根据Jin et al. 的研究,我们使用迭代目标增强(Iterative Target Augmentation, ITA)算法来解决这个优化问题。因为最初的算法目的是计算离散属性,所以我们将其改为适用于连续值(亲和力指标)的版本,具体细节见附录B。在这个过程中,我们舍弃了所有PPL低于10的结构。可以观察到我们的模型能够隐式地学习到净电荷、基序、重复氨基酸,所以我们不需要显示地使用声明。
结果
见表2中间的表格,MEAN在设计亲和力更高的抗体上表现比其它方法都要优越。这进一步证明了显示对不同组分界面建模的优点,此外,为了更好解释我们的结果,我们提供了随机突变后CDRs的ΔΔG预测值(表示为Random),在附录C中详细解释了这一结果。我们也在图3(B)中提供了一个可视化的例子,表明MEAN确实可以设计一个亲和力更高的新的CDR-H3序列/结构。
给定对接模板的CDR-H3设计
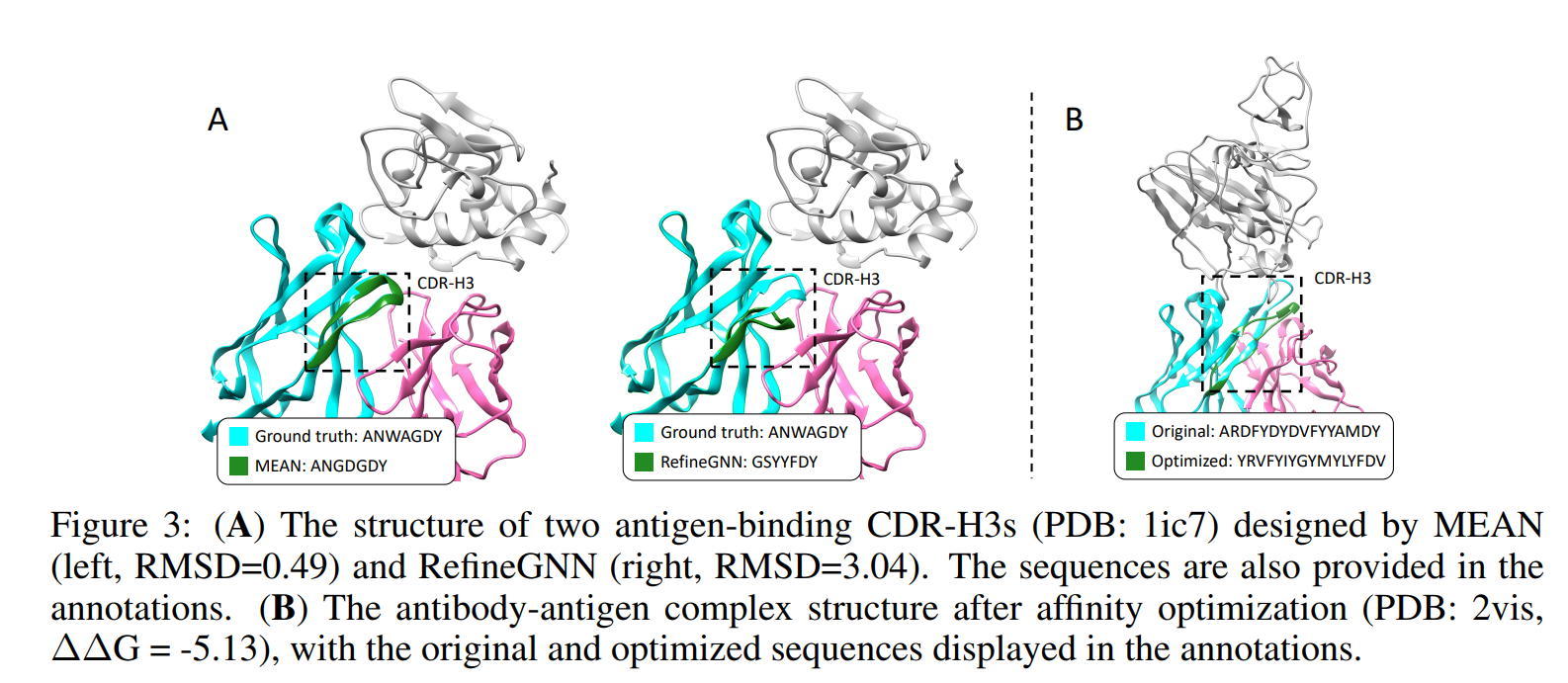
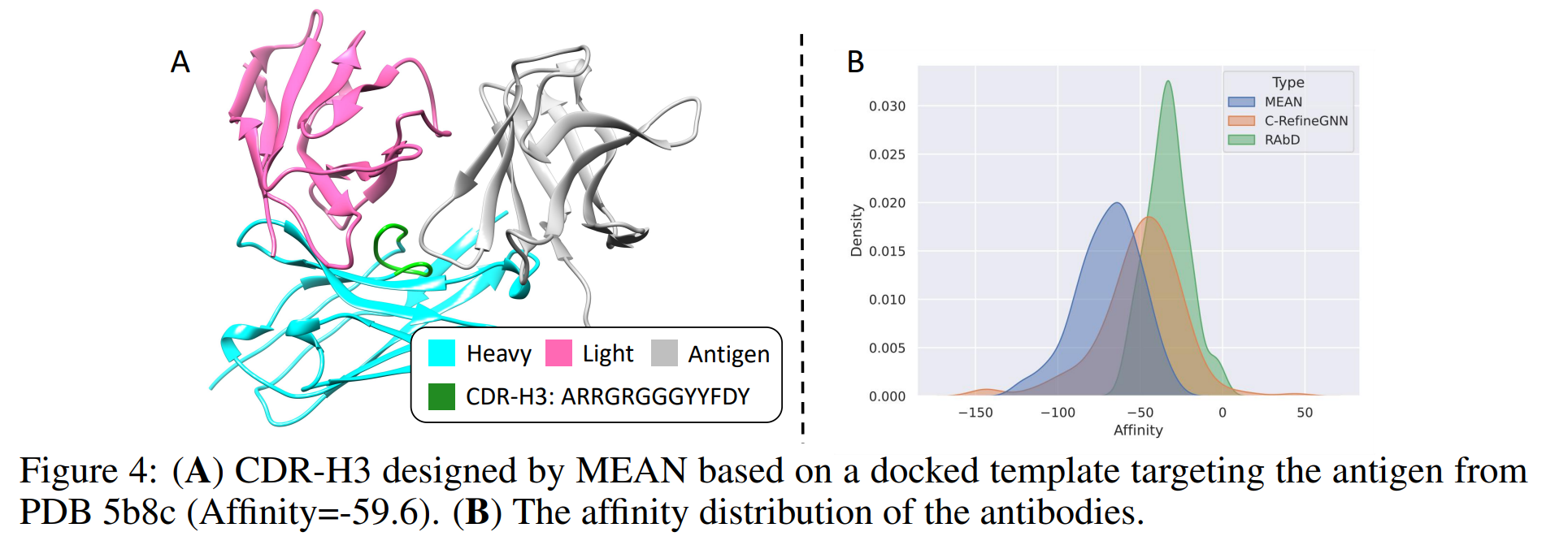
我们进一步提供了一个使用我们模型的流程,即结合复合体未知的情况。具体地,对于RAbD中的抗原,我们旨在生成与其结合亲和力尽可能高的抗体。为了这个目的,我们首先从数据库中选择一个抗体,并去除其CDR-H3,然后再使用HDOCK来对接到目标抗原,以此来获得一个抗原抗体复合体。有了这个模板复合体,再使用我们的模型生成抗原结合的CDR-H3s(如4.2)。为了缓解对接过程的不准取性,我们为每个抗原构建了10个模板,并保留了下一次生成中打分最高的一个。首先,我们使用OpenMM和Rosetta来优化生成的模型,然后使用Rosetta的能量函数来测量结合亲和力。图4中展示了MEAN生成抗体以及C-RefineGNN和RAbD数据库中原始的亲和力分布(图4 B)。显然,MEAN设计的抗体预测的亲和力要更高,图4 A中给出了一个紧密结合的例子。
分析 Analysis
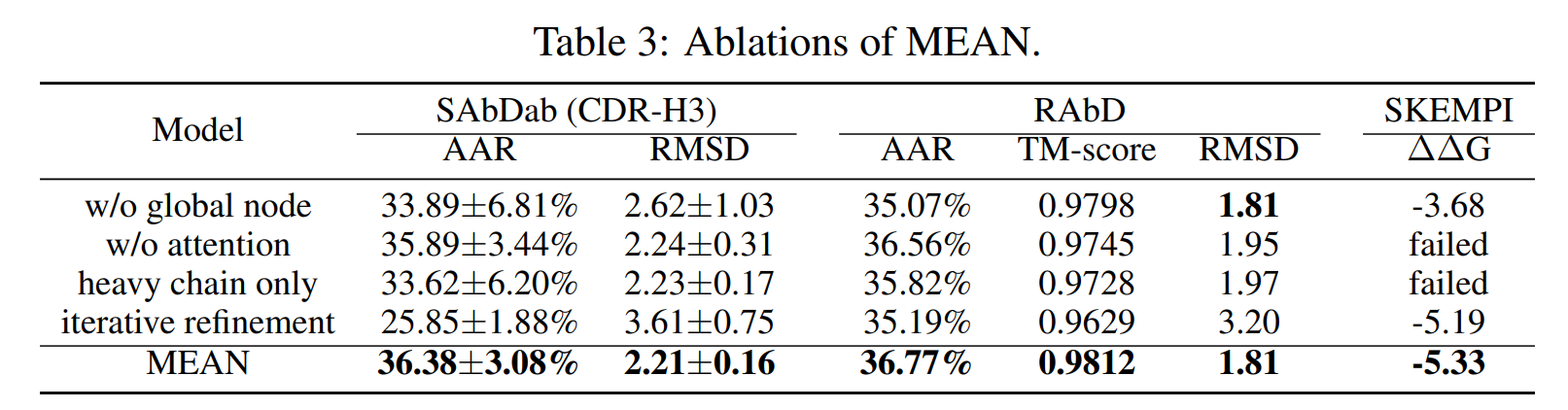
我们测试了是否MEAN中每个步骤都是不可缺少的。表3表明了去除全局节点和注意力机制会导致性能变差。由于全局节点抗原在元件内和元件之间传递信息,而注意力模块关注于不同元件界面的局部信息,所以删除这两个部分都会导致性能变差。实际上,注意力模块也为两两氨基酸之间互作的重要性提供了解释,见附录I。此外,表3表明只是用重链信息也会导致性能变差,而且不能够再亲和力优化上取得SOTA,这进一步说明了加入完整结构信息的重要性。此外,我们实现了一个MEAN的变体,将逐步完全解码改为了自回归解码,性能随之变差,而且模型效率由于迭代次数的增加也随之变慢。如表2(右)所示,我们的方法随着CDR序列的长度,推理速度能够提升2-5倍。我们也分析了MEAN的复杂度,插入随机性,以及逐步解码过程,分别见附录G、H和J。
结论
本研究将抗体设计工作构想成将抗体抗原复合物的整个背景作为输入,以通用 CDR 为输出。提出了多通道等变注意网络(MEAN)来识别和编码不同链内部和链间的局部信息和全局信息。还提出了渐进式全镜头解码策略,以实现更高效、更精确的生成。作者的模型在1D 序列和 3D 结构的学习、CDR-H3抗原结合设计和亲和力优化三个方面大幅度领先于基线。